NetPhorce Plotting Demonstration
Source:vignettes/rMD_V2_TwoConditionDemonstrationWithPlots.rmd
rMD_V2_TwoConditionDemonstrationWithPlots.rmd1.0 Process the Data
## Loading One Condition Data
data("twoConditionsExample", package = "NetPhorce")
## ## Identify the Key Columns
identifiedCols <- confirmColumnNames(rawMaxQuant = twoConditionsExample,
positionCol = "Position",
reverseCol = "Reverse",
localizationProbCol = "Localization prob",
potentialContaminationCol = "Potential contaminant",
aminoAcidCol = "Amino acid",
uniqueIDCol = "Protein",
seqWindowIDCol = "Sequence window",
fastaIDCol = "Fasta headers")
## Identify the Intensity Columns with Condition, Time Point and Replication Information
intensityCols <- confirmIntensityColumns(rawMaxQuant = twoConditionsExample,
intensityPattern = "con_time_rep",
verbose = TRUE)
## Process the data based on the identified columns
netPhorceData <- processData(rawMaxQuant = twoConditionsExample,
processedColNames = identifiedCols,
processedIntensity = intensityCols,
minReplication = 3,
minLocalProb = 0.75)
#>
#> Number of Conditions Found: 2. Undergoing a time consuming step, please wait...
#>
#> Complete.
## Validating the Kinase/Phosphatase Information
netPhorceData <- validateKinaseTable(netPhorceData = netPhorceData,
defaultKinaseTable = TRUE,
abbrev = "Ath")
#> Kinase and Phosphatase Matching Table:
## Regulation Validation based on user inputs
netPhorceData <- regulationCheck(netPhorceData = netPhorceData,
upReg = 0.25,
downReg = 0.25,
absMinThreshold = 0.1,
qValueCutOff = 0.05,
verbose = TRUE)
## Network Analysis
netPhorceData <- networkAnalysis(netPhorceData = netPhorceData,
requestPlotData = TRUE)
#>
#> Undergoing a time consuming step, please wait...2.1 Distribution Plot
2.1.1 Static Version
plotDistribution(netPhorceData = netPhorceData,
condition = "Col0",
plotly = FALSE)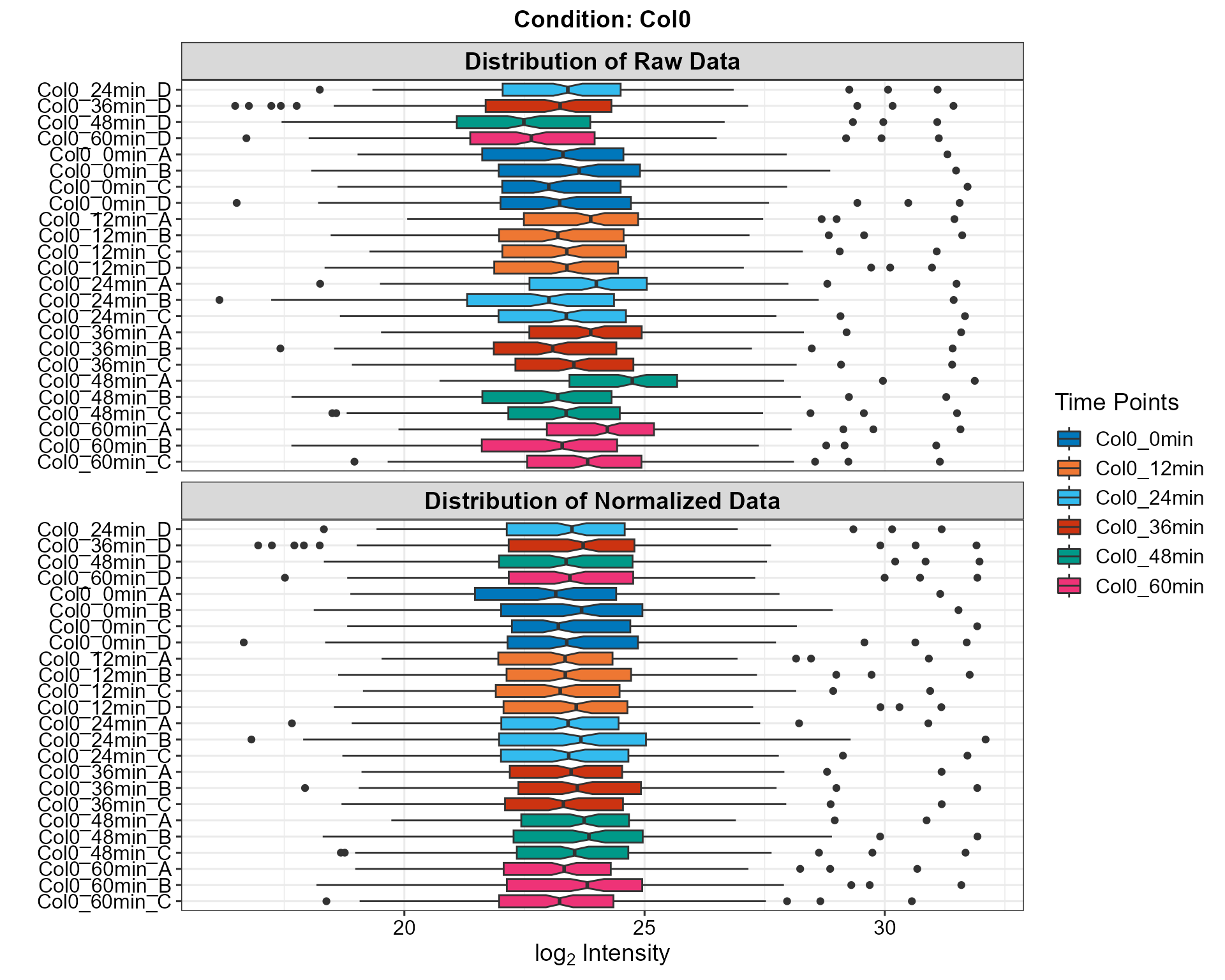
2.1.2 Interactive Version
plotDistribution(netPhorceData = netPhorceData,
condition = "Col0",
plotly = TRUE)2.2 Histogram and Boxplot
2.2.1 Histogram and Boxplot Side-by-Side Plot
plotHistBox(netPhorceData = netPhorceData,
condition = "tot3",
histogram = TRUE,
boxplot = TRUE)
2.2.2 Boxplot Only
plotHistBox(netPhorceData = netPhorceData,
condition = "tot3",
histogram = FALSE,
boxplot = TRUE)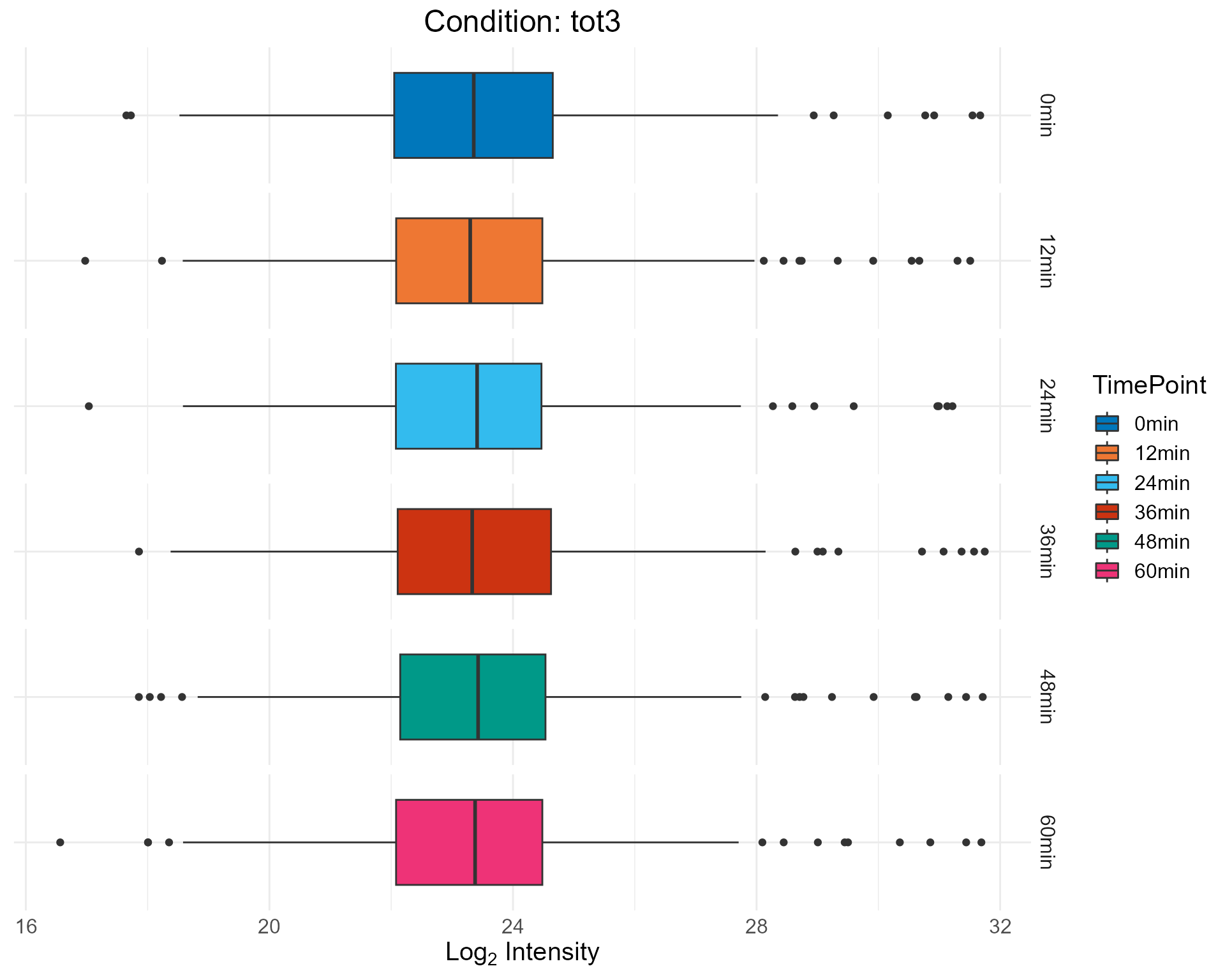
2.2.3 Histogram Only
plotHistBox(netPhorceData = netPhorceData,
condition = "tot3",
histogram = TRUE,
boxplot = FALSE)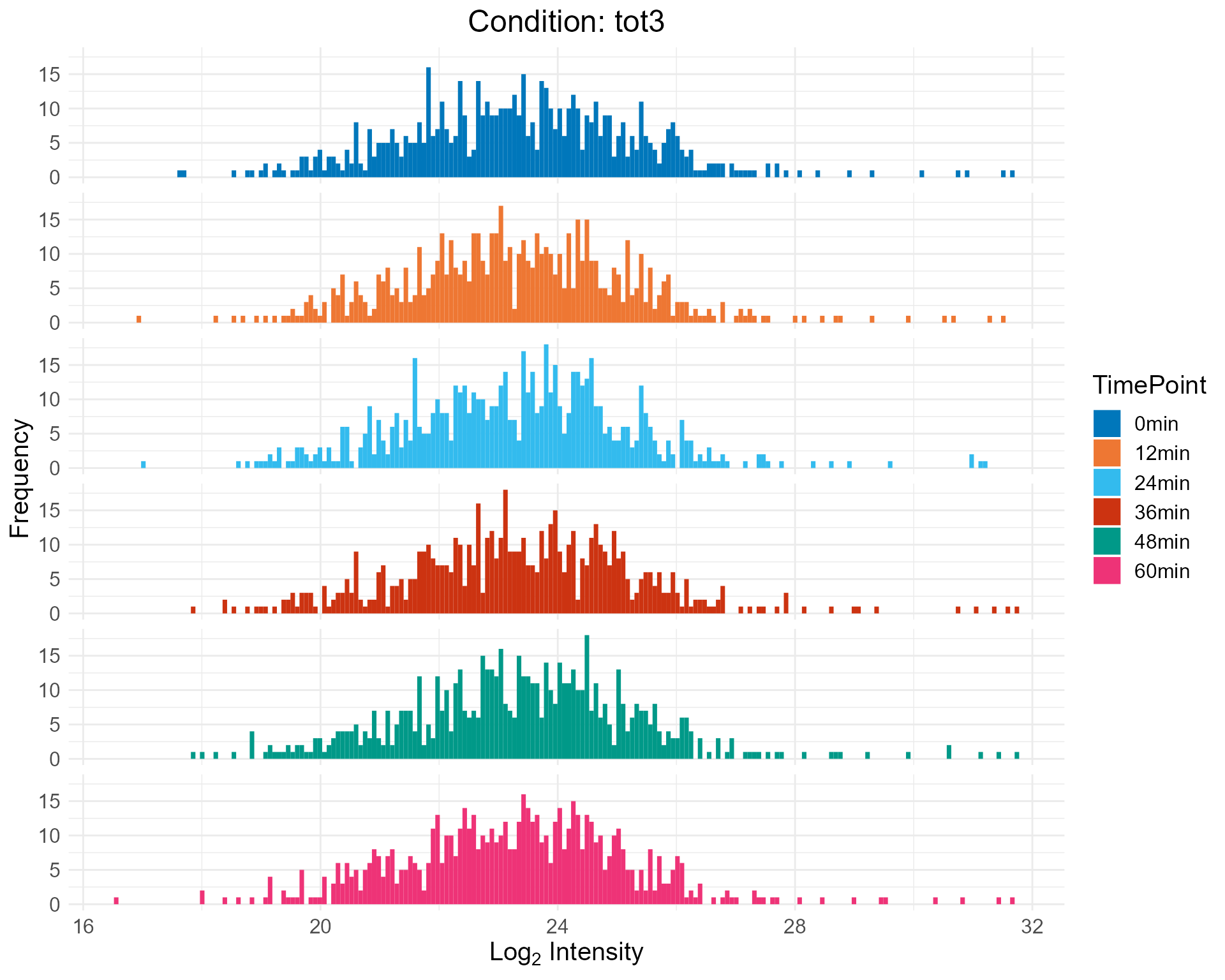
2.3 PCA Plot
2.3.1 Static Version
plotPCA(netPhorceData = netPhorceData,
condition = "Col0",
normalized = FALSE,
plotly = FALSE)
2.3.2 Interactive Version
plotPCA(netPhorceData = netPhorceData,
condition = "tot3",
normalized = TRUE,
plotly = TRUE)2.4 Heatmap based on Unique IDs
2.4.1 Identify Unique IDs
uniqueIDs <-
findUniqueIDs(netPhorceData = netPhorceData,
uniqueIDList = c(
# Significant Set Examples:
"AT1G13030.1", "AT1G13360.3", "AT1G42550.1",
# Unique/Abscence Set Examples
"AT1G17280.9", "AT1G22310.2", "AT1G23890.2"
),
verbose = TRUE)
#> All 6 provided unqiue IDs are matched to existing Protein IDs2.4.2 Heatmap Based on Found Unique IDs - Significant Set Interactive Version
plotUniqueIDsHeatmaps(netPhorceData = netPhorceData,
foundUniqueIDs = uniqueIDs,
heatmapType = "Significant",
minQVal = 0.05,
plotly = TRUE)
#> Joining with `by = join_by(UniqueID)`
#> ..cutHeight not given, setting it to 18.3 ===> 99% of the (truncated) height range in dendro.
#> cutHeight set too low: no merges below the cut.2.4.2 Heatmap Based on Found Unique IDs - Absence/Presence Set Static Version
plotUniqueIDsHeatmaps(netPhorceData = netPhorceData,
foundUniqueIDs = uniqueIDs,
heatmapType = "AbsencePresence",
plotly = FALSE)
#> ..cutHeight not given, setting it to 84.2 ===> 99% of the (truncated) height range in dendro.
#> ..done.
2.4 Heatmap based on Cluster IDs
2.4.1 Identify Cluster IDs from Significant Set
clusterIDs_Sig <-
findClusters(netPhorceData = netPhorceData,
clusterIDs = c(0),
heatmapType = "Significant",
minQVal = 0.1,
verbose = TRUE)
#> Joining with `by = join_by(UniqueID)`
#> ..cutHeight not given, setting it to 33.7 ===> 99% of the (truncated) height range in dendro.
#> cutHeight set too low: no merges below the cut.
#> All 1 cluster are matched with provided q-value cut-off: 0.12.4.2 Identify Cluster IDs from AbsencePresence Set
Note, you do not need to supply minQVal for Abscence Presence Heatmap
clusterIDs_AbsPrs <-
findClusters(netPhorceData = netPhorceData,
clusterIDs = c(1),
minQVal = 0.1,
heatmapType = "AbsencePresence",
verbose = TRUE)
#> ..cutHeight not given, setting it to 84.2 ===> 99% of the (truncated) height range in dendro.
#> ..done.
#> All 1 cluster are matched with provided q-value cut-off: 0.12.4.3 Heatmap Based on Found Unique IDs - Significant Set Interactive Version
plotClustersHeatmap(netPhorceData = netPhorceData,
foundClusterIDs = clusterIDs_Sig,
plotly = TRUE)
#> Joining with `by = join_by(UniqueID)`
#> ..cutHeight not given, setting it to 33.7 ===> 99% of the (truncated) height range in dendro.
#> cutHeight set too low: no merges below the cut.2.4.4 Heatmap Based on Found Unique IDs - Significant Set Static Version
plotClustersHeatmap(netPhorceData = netPhorceData,
foundClusterIDs = clusterIDs_AbsPrs,
plotly = FALSE)
#> ..cutHeight not given, setting it to 84.2 ===> 99% of the (truncated) height range in dendro.
#> ..done.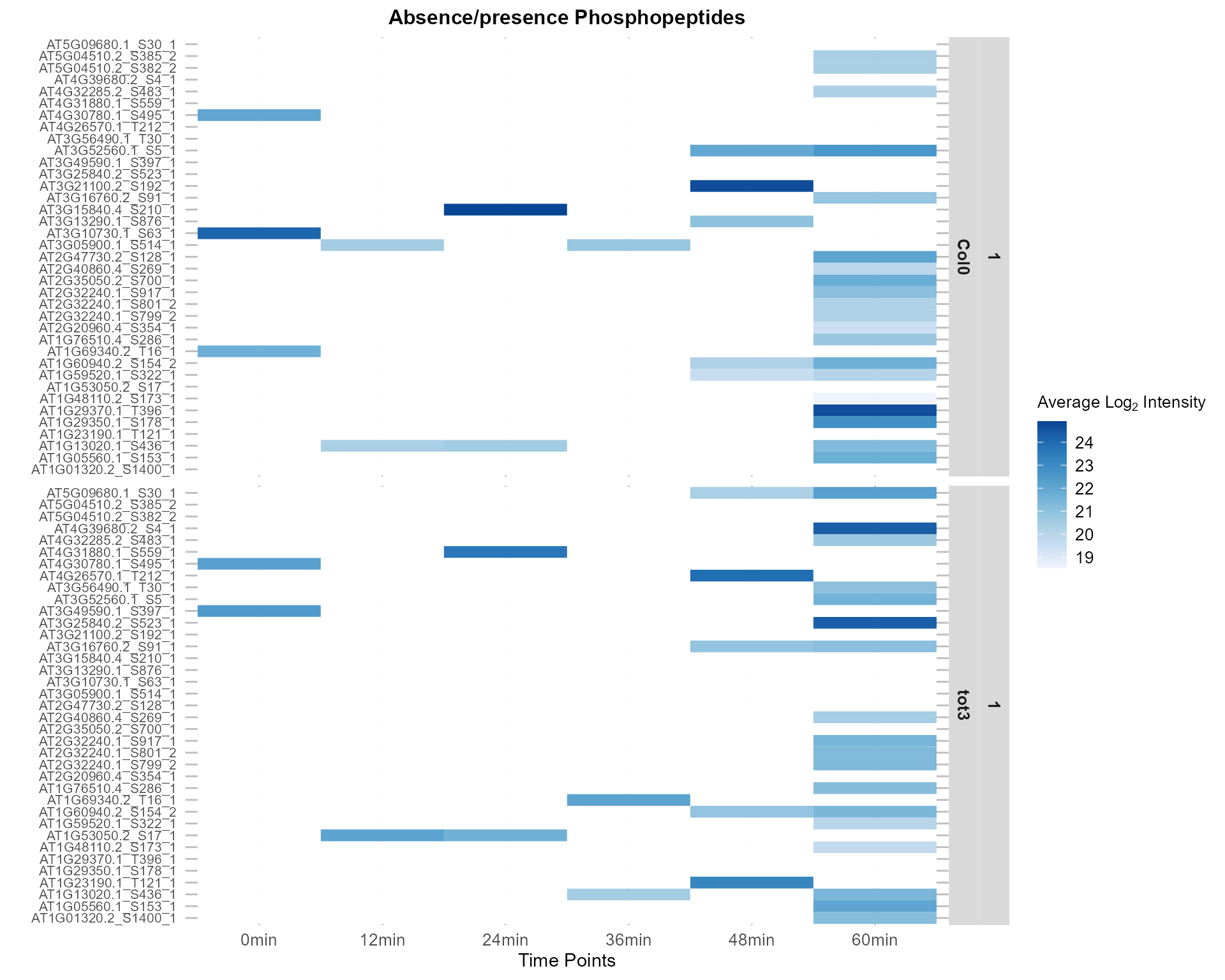
2.6 Single or Multiple Peptides Plot
2.6.1 Extract Peptide ID
peptideIDs <-
findPeptideIDs(netPhorceData = netPhorceData,
peptideIDList = c("AT1G01320.2_S1349_1", "AT1G05560.1_S153_1", "AT1G01320.2_S149_1"))
#> All 3 provided phosphopeptide IDs are matched to existing phosphopeptide IDs2.6.2 Single Peptide Plot - Interactive Version
plotSinglePeptide(netPhorceData = netPhorceData,
foundPepetidesIDs = peptideIDs,
plotAll = FALSE,
plotly = TRUE)
#> Multiple Phosphopeptide IDs provided, only plotting the first one. Please
#> use plotAll if you want to plot all the found phosphopeptide IDS.2.6.3 Single Peptide Plot - Static Version
plotSinglePeptide(netPhorceData = netPhorceData,
foundPepetidesIDs = peptideIDs,
plotAll = FALSE,
plotly = FALSE)
#> Multiple Phosphopeptide IDs provided, only plotting the first one. Please
#> use plotAll if you want to plot all the found phosphopeptide IDS.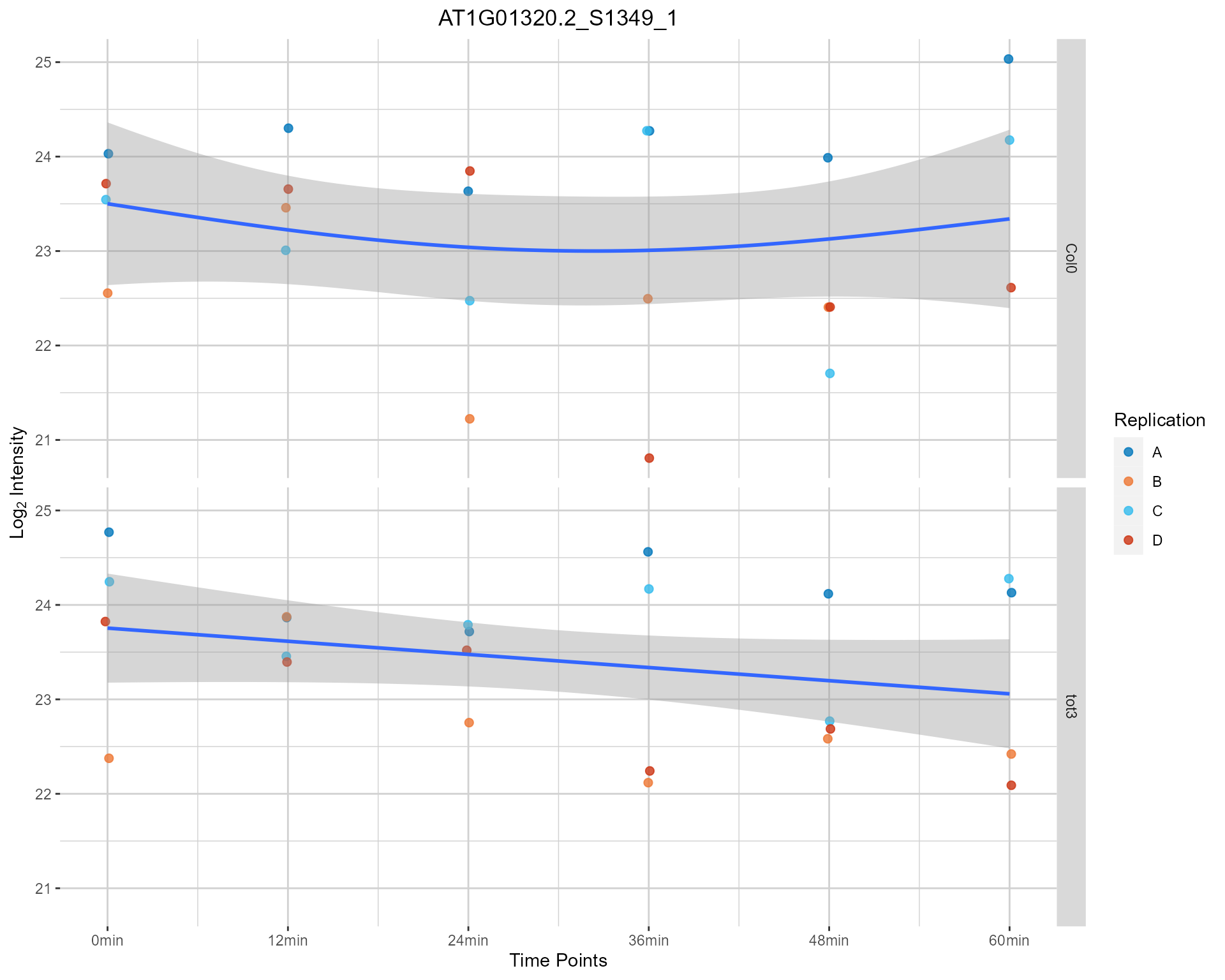
2.6.4 Multiple Peptides Plot - Interactive Version
plotMultiPeptides(netPhorceData = netPhorceData,
foundPepetidesIDs = peptideIDs,
condition = "Col0",
plotly = TRUE)2.6.5 Multiple Peptides Plot - Static Version
plotMultiPeptides(netPhorceData = netPhorceData,
foundPepetidesIDs = peptideIDs,
condition = "tot3",
plotly = FALSE)
3.0 Regulation Plot
3.1 Regulation Plot - Interactive Version
plotRegulation(netPhorceData = netPhorceData ,
condition = "Col0",
plotly = TRUE)3.2 Regulation Plot - Static Version
plotRegulation(netPhorceData = netPhorceData ,
condition = "tot3",
plotly = FALSE)
4.0 Network
plotNetPhorce(netPhorceData = netPhorceData,
condition = "tot3",
FASTADescription = TRUE)